主な研究テーマ
私たちは、(1)自然選択がゲノムに与える影響、(2)集団の起源・移住・混血過程、(3)感染症とヒトの遺伝的適応、(4)オセアニア地域集団における体型・脂質代謝関連多型、(5)ゲノムインフォマティクス、(6)理論集団遺伝学・理論生物学について研究しています。
自然選択がゲノムに与える影響
-正の自然選択-
分子進化の大部分は中立的な変異の蓄積に起因しますが、生物進化の過程で表現型に大きな影響を及ぼした変異の中には正の自然選択が作用したものが多いと考えられます。私たちは、遺伝子配列解析・多型解析・統計解析・集団遺伝学的解析(コンピュータシミュレーション)によって、正の自然選択が作用して進化してきた遺伝子を同定し、その分子進化のメカニズムを解明したいと考えています。特に注目しているのは、有利なアリルが未だ固定には至っていない最近の正の自然選択についてです。連鎖不平衡を指標とし、最近の正の自然選択のターゲットとなった変異の選択強度や誕生時期を推定することができます。また、関連解析などによって、正の自然選択の対象となった表現型を知ることもできます。Coriell社より購入したHapMapサンプルを用いて、HapMapプロジェクトや1000ゲノムプロジェクトでは調べられていない(データベースに登録されていない)多型の解析を行い、周辺多型との連鎖不平衡の程度から、最近の正の自然選択が作用してきたアリルを同定する試みも行っています。
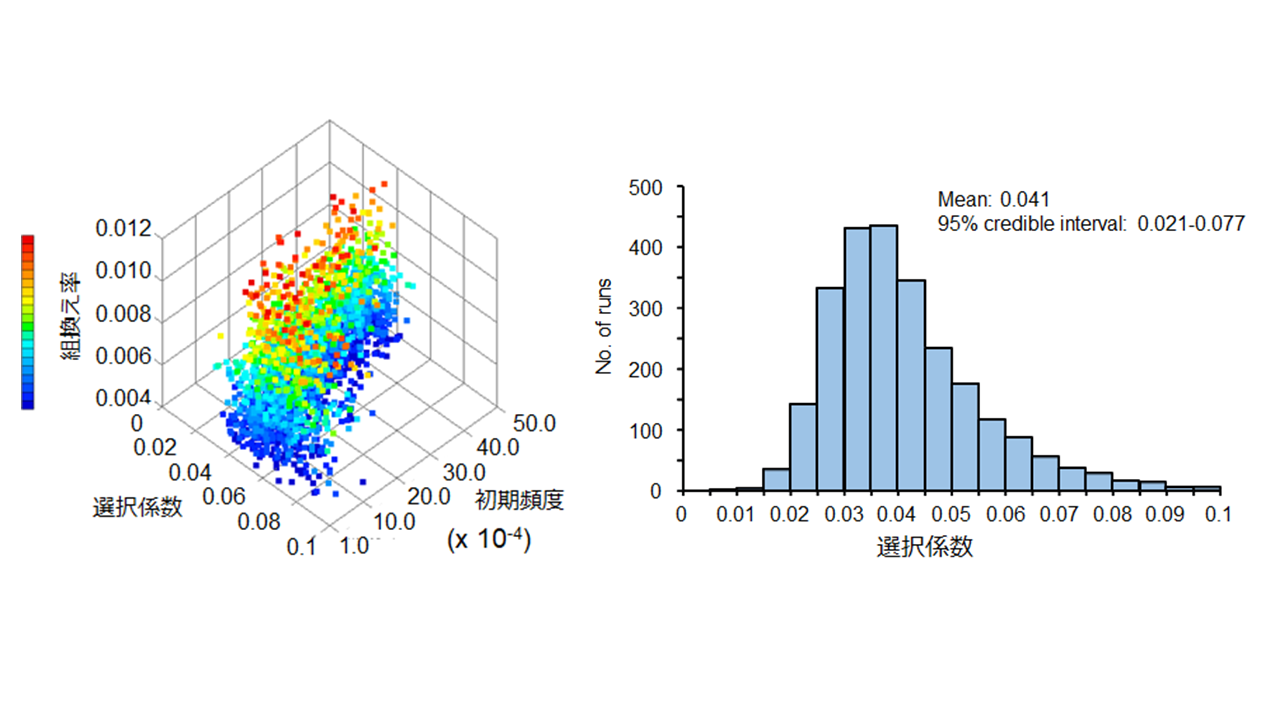
日本人集団においてHLA-DPB1*0401アリルに作用した最近の自然選択の解析結果。棄却サンプリング法を用いた近似ベイズ計算により推定した、初期頻度、組み換え率、選択係数の組合せ(左)と選択係数の事後分布(右)。HLA-DPB1*0401アリルが乗るHLAハプロタイプは渡来人由来であり、弥生時代以降に強い正の自然選択(他のアリルより4%程度有利)が作用したことが示唆された。
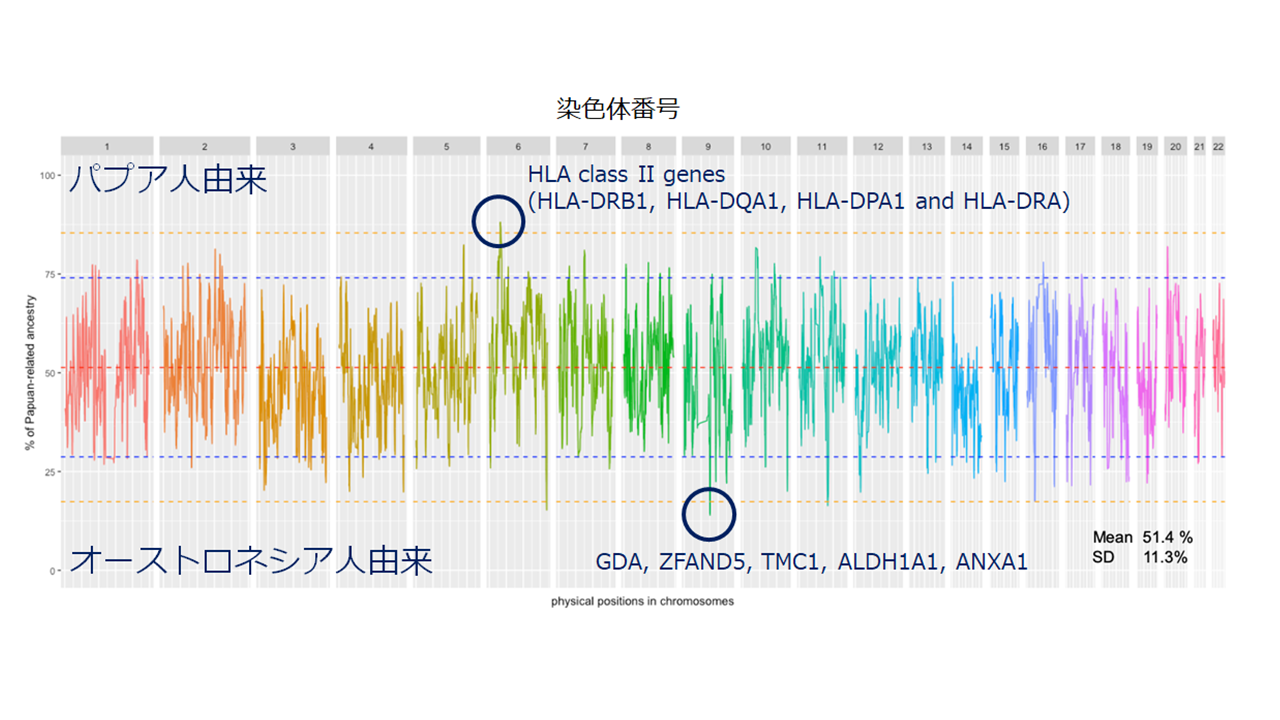
オーストロネシア人(ソロモン諸島民)を対象にした染色体領域ごとの由来解析結果。ソロモン諸島民はパプア人と台湾起源のオーストロネシア人の混血集団である。ELAI解析の結果、ソロモン人のゲノム成分のほぼ半分がパプア人由来であり、残り半分がオーストロネシア人由来と推定された(図の赤破線)。パプア人由来成分が多く観察されるのは6番染色体のHLAクラスII遺伝子領域、オーストロネシア人由来成分が多く観察されるのは9番染色体のANXA1遺伝子を含む領域であった。オーストロネシア人がメラネシアに到達して現地のパプア人と混血した際に、パプア人がもっていたHLAクラスIIアリルが地域特異的適応に寄与した(メラネシア地域の感染症に対して抵抗性を示すHLAクラスIIアリルが自然選択により頻度増加した)可能性がある。
-HLA遺伝子に作用する超優性選択-
ヒトゲノム中で最も強い自然選択が作用してきたのはHLA(ヒトのMHC)遺伝子です。HLA分子の主要な機能は病原体由来ペプチドをTCRに提示することです。ヘテロ接合の個体は2種類のHLA分子を発現するため、ホモ接合体よりも多くの種類のペプチドをTCRに提示します。多種類のペプチドを提示できるということは、より多くの病原体に対抗できることを意味します。そのため、ヘテロ接合体はホモ接合体よりも生存上有利になります(このような自然選択を超優性選択といいます)。アリル数が増えるとヘテロ接合体頻度も増えるため、自然選択の効果により数千を超える膨大な種類のアリルがヒト集団中に維持されています。特に興味深いのは、他のHLAアリルよりもチンパンジーのアリルに配列が似ているヒトのHLAアリルが存在することです。つまり、多型が種を超えて維持されているのです。私たちは、そのような種を超えたHLA多型を調べ、一般的な遺伝子やHLA領域以外のゲノム領域の解析からはわからないヒト祖先集団の進化史について研究しています。また、マラリア、デング熱、HBV、HIVなどに感染した個体のHLA遺伝子を調べ、現在作用する自然選択についても研究しています。
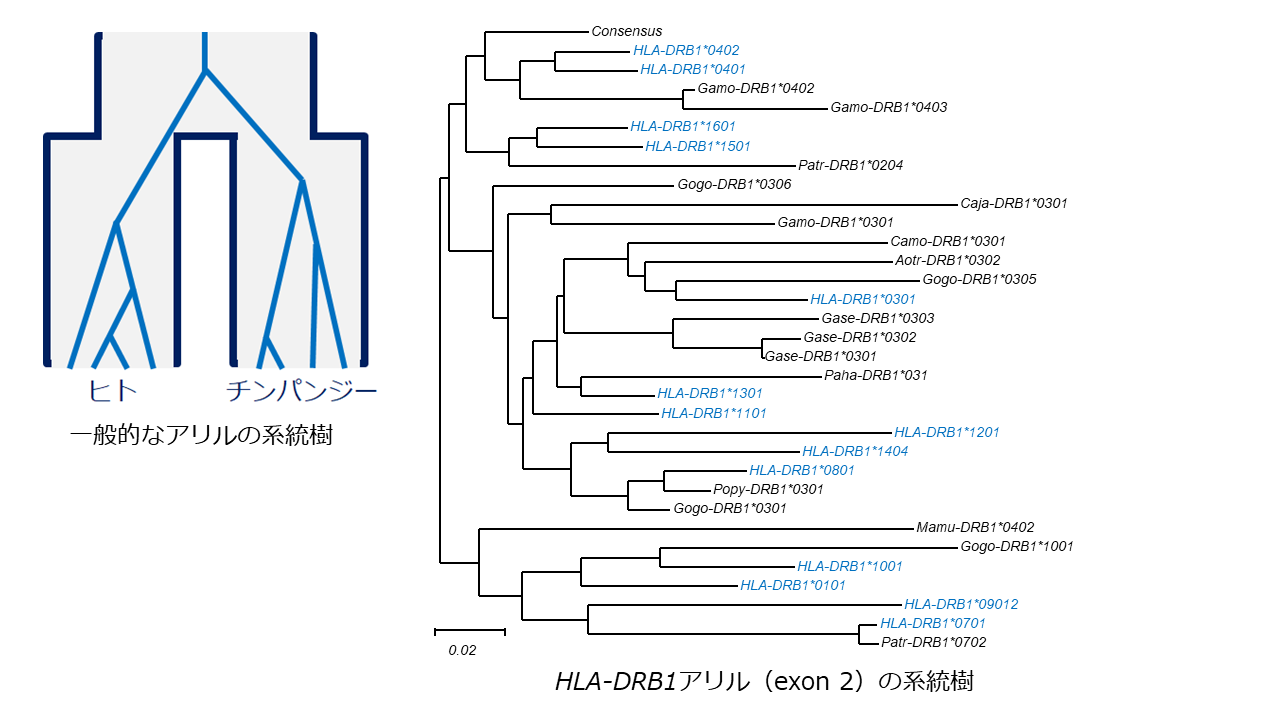
一般的な遺伝子座で観察されるアリル系統樹の例(左)とDRB1アリルの系統樹(右)。ヒトやチンパンジーの中立な遺伝子座(一般的な遺伝子座)では、種内に遺伝子コピーの共通祖先が存在するため、ヒトのアリル同士、チンパンジーのアリル同士は配列が似ている。一方、HLA遺伝子座では、チンパンジーやゴリラのアリルとの方が似ているヒトのアリルが存在する。すなわち、種分岐以前に誕生したHLAアリル(多型)が現在まで受け継がれている(これを、「種を超えた多型」という)。
集団の起源・移住・混血過程
-日本人集団の形成過程-
私たちは、日本列島人の起源や日本人集団の形成過程について調べています。アイヌ人、琉球人、本土日本人(アイヌ人と琉球人を除く日本人)のゲノムワイドSNP解析によって、本土日本人と韓国人とは近い関係にあることが分かりました。これは、弥生時代に朝鮮半島経由で日本列島へ移住した人々(渡来人)の子孫が現代本土日本人の主体であることを反映していると思われます。
また、Y染色体の解析から、縄文時代晩期から弥生時代にかけて、縄文人(男性)集団の急激な人口減少と人口増加が起きていたことがわかりました。現在は、47都道府県を対象にしたゲノムワイドSNP解析によって、日本列島内での縄文人と渡来人の混血過程について調べています。
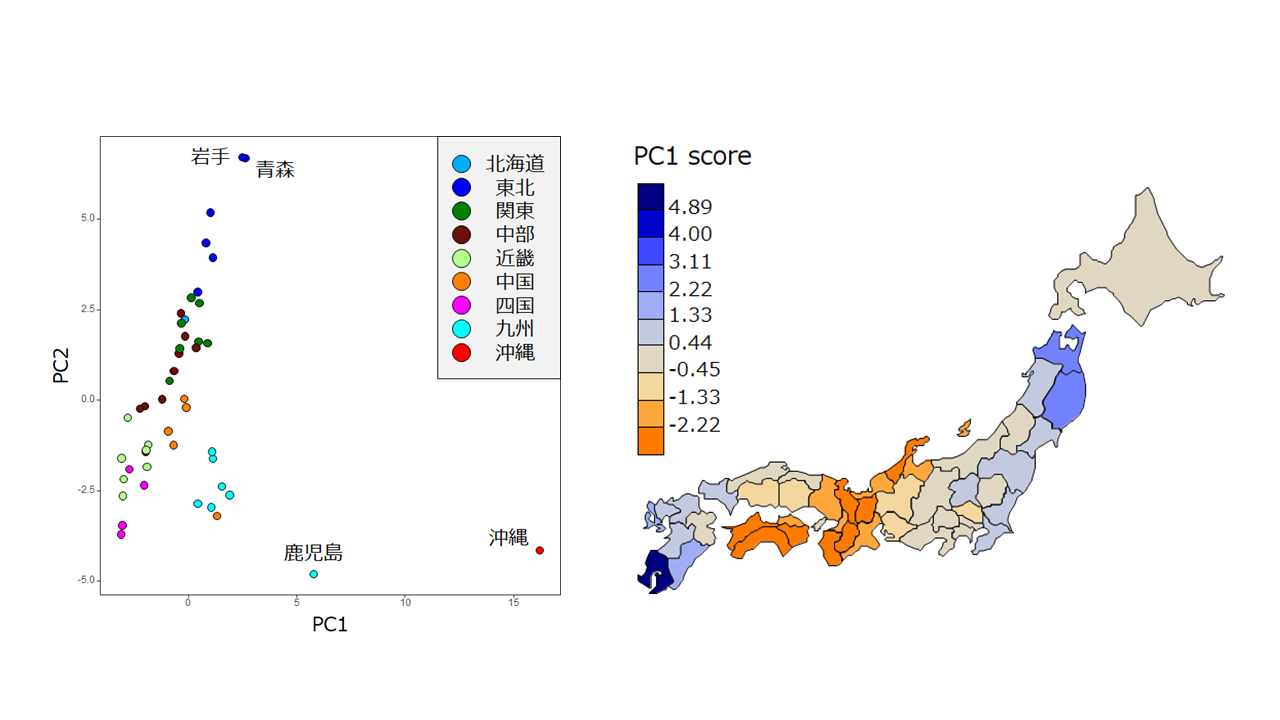
ゲノムワイドSNP頻度データを用いた47都道府県に対する主成分分析の結果(左)と第一主成分(PC1)の値によって都道府県を色分けした図(右)。右図では、沖縄県は除いてある。青色が濃いほど沖縄県と遺伝的に近く、オレンジ色が濃いほど遠い。沖縄県に近い県は東北地方と九州地方に多く、遠い県は近畿地方と四国地方に多い。また、近畿地方や四国地方の県は遺伝的に中国・漢民族と近かった。琉球人と縄文人は遺伝的に近縁であり、渡来人は現代の中国人と祖先を共有していると考えられることから、渡来人の遺伝成分は近畿地方や四国地方により多く残っている思われる。大陸から来た渡来人の遺伝成分の割合が日本列島の中央部で高いことは興味深い。
-オセアニア集団の移住・拡散-
南太平洋に位置するオセアニアは、地理学的にメラネシア、ミクロネシア、ポリネシアに、言語学的には非オーストロネシア語族とオーストロネシア語族とに分類されます。人類学的、考古学的、言語学的研究などにより、人類のオセアニア地域へ大規模な移住が、過去2回あったことが分かっています。今から約5万年前には、非オーストロネシア語を話す集団が、サフル大陸(現在のオーストラリア大陸とタスマニア島、ニューギニア島の陸つづき)に到達したと考えられています(オセアニアへの第一の移住)。その後、サフル大陸が現在とほぼ同じ地形になった約5000年前、オーストロネシア語を話す集団が、東南アジアからニューギニア島北側を西から東へ移住したと考えられています(オセアニアへの第二の移住)。第二の移住者はとても速く拡散しましたが、未だ正確な拡散ルート、第一の移住者と第二の移住者との遺伝的近縁関係は明らかにされていません。私たちは、フィールドワークによってオセアニア地域集団(パプアニューギニア、ソロモン、トンガ)の人々からDNA試料を提供していただき、各種遺伝マーカー(HLA、ABO、mtDNAなど)やゲノムワイドSNPデータの解析を通して、オセアニア集団の移住・拡散ルートの解明を目指しています。
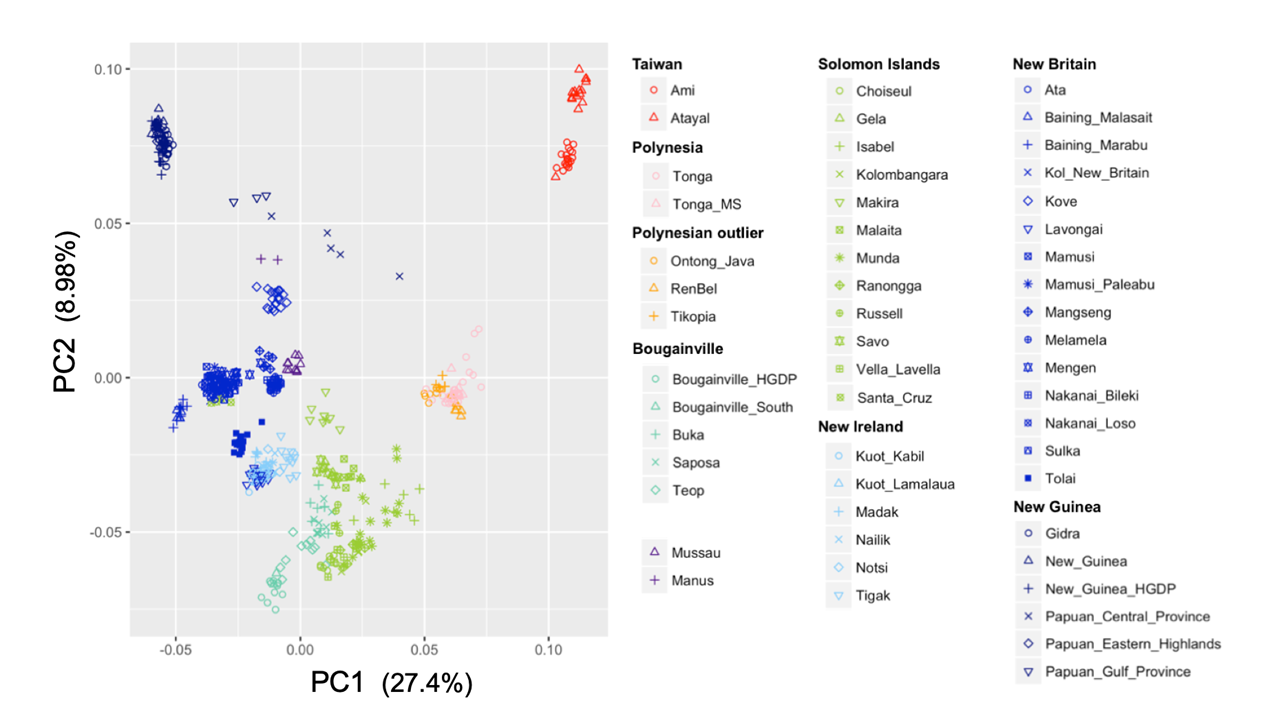
ゲノムワイドSNPデータを用いたオセアニア・東南アジア集団に対する主成分分析の結果。第一主成分(PC1)に着目すると、左上のパプア人と右上の台湾人の中間にオセアニアのオーストロネシア人が位置しており、オセアニア地域で第一の移住者と第二の移住者が混血したことがわかる。
感染症とヒトの遺伝的適応
-新型コロナウイルス感染症-
新型コロナウイルス感染症(国際正式名称:COVID-19)は、SARSコロナウイルス2 (SARS-CoV-2) がヒトに感染することによって発症する感染症です。2019年に発生すると、強力な感染力によって瞬く間に全世界に感染が拡大しました。
われわれ人類は、新たな感染症に対して適応的に働くような既存の多型を保有しているのでしょうか。
私たちは、日本人の新型コロナウイルス感染患者を対象として、新型コロナウイルス感染やその重症化と関連するヒト遺伝子多型の解析を行っています。
また、SARSコロナウイルス2ゲノムの分子進化学的解析によって、SARSコロナウイルス2が高い感染力を獲得する進化過程(変異株の誕生過程)についても調べています。
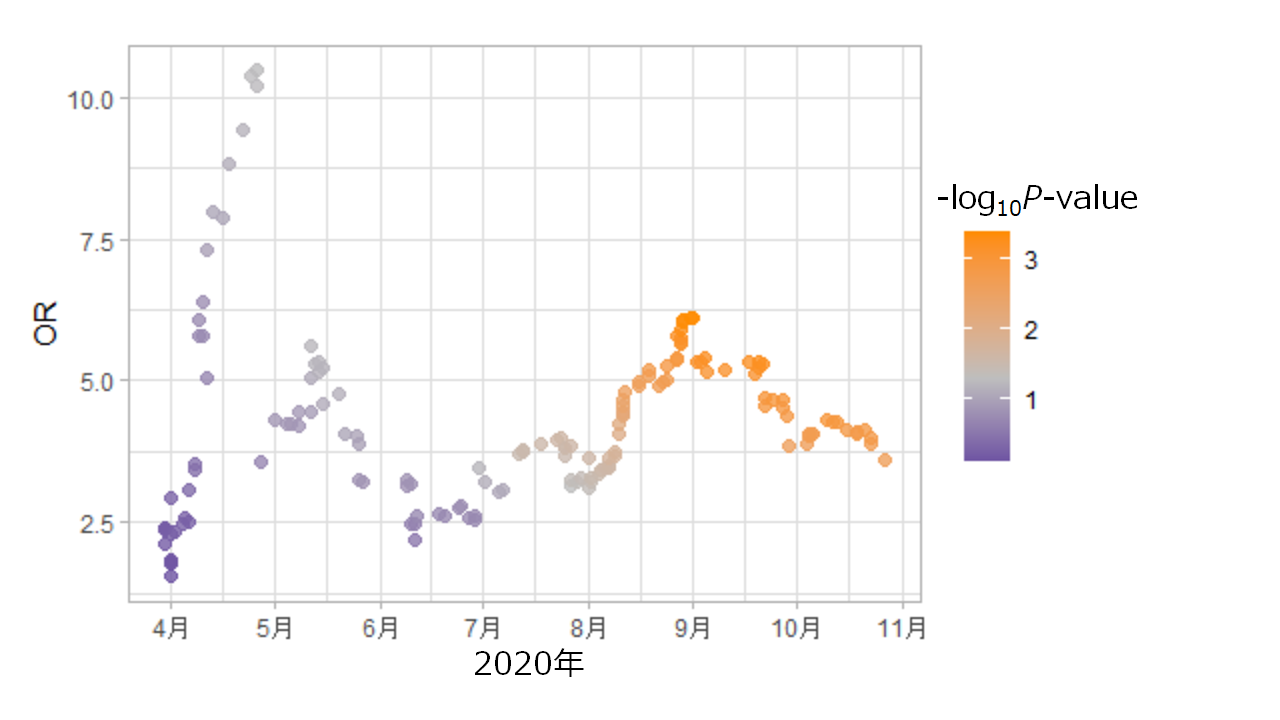
新型コロナウイルス感染症重症化とHLA-DRB1*09:01アリルの関連。 2020年4月以降に感染した新型コロナウイルス患者を累積してオッズ比(OR)と関連P値の変化を図示した。HLAアリルの関連は、SARSコロナウイルス2の進化(コードするタンパク質のアミノ酸配列)に影響を受ける可能性があるため、今後も注視していく必要がある。
-マラリア-
マラリアは熱帯・亜熱帯地域に生息するハマダラ蚊属の蚊の吸血によって媒介する感染症で、熱帯熱マラリア・四日熱マラリア・三日熱マラリア・卵型マラリアの4種類があります。その中でも熱帯熱マラリアは症状が重く、重症化すると死亡することもまれではありません。私たちは、タイ北西部に居住するタイ人マラリア患者を対象として、脳性マラリアなどのマラリア重症化に対する感受性または抵抗性を示す遺伝子多型の検出とその進化学的解析を行っています。また、マラリア患者(ホスト)とその患者に感染している熱帯熱マラリア原虫(病原体)を対象に、マラリア感染の成立・維持の過程において、直接的に作用しあうヒト側分子と原虫側分子をコードする遺伝子を解析して、ヒトと原虫の遺伝子多型間の相互作用や、多型や遺伝子に作用する正の自然選択の検出を試みています。

タイ(タック県)での熱帯熱マラリア感染調査。
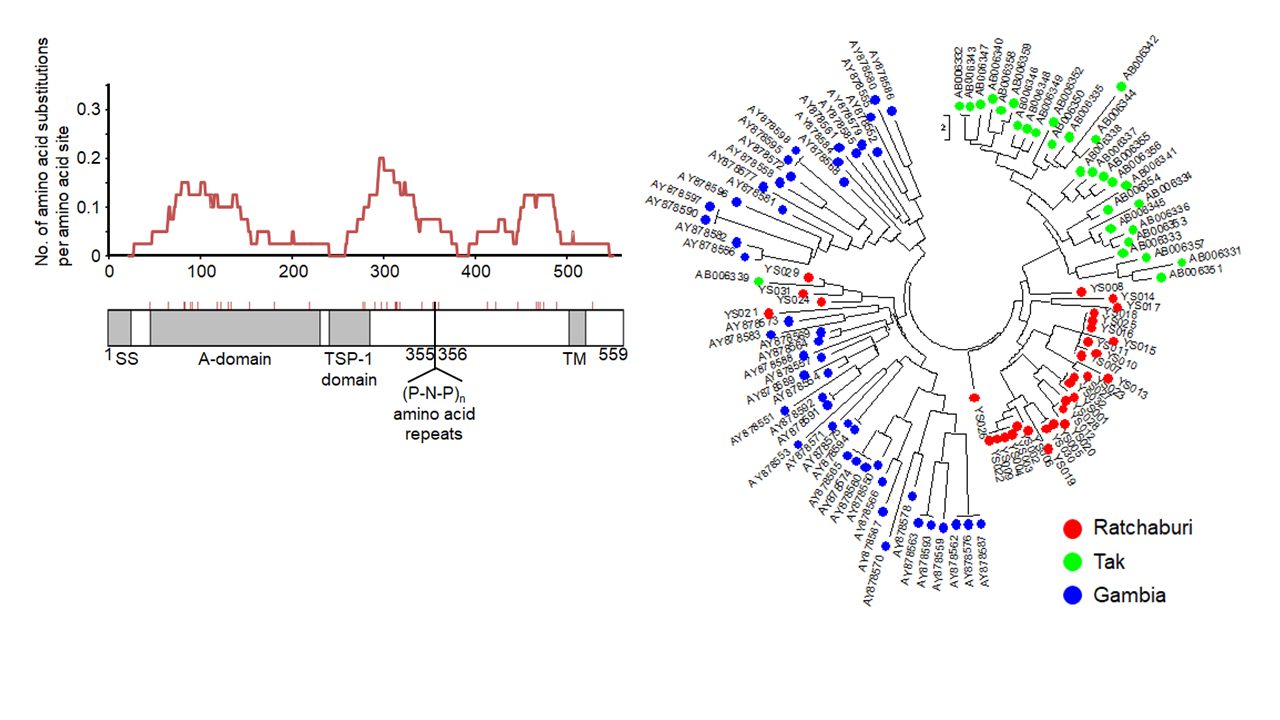
熱帯熱マラリア原虫のTRAP遺伝子の多様性解析。タイのTak県で採取した熱帯熱マラリア原虫(熱帯熱マラリア患者から抽出)で観察されたTRAP遺伝子中のアミノ酸置換密度解析(左図)。タイのRatchaburi県およびアフリカ(ガンビア)の熱帯熱マラリア原虫を含めた分子系統樹(右図)。地域特異的に多様性が高く、各地域で多様化選択(diversifying selection)が作用していることがわかる。
-デング熱-
デングウイルスの感染によって引き起こされるデング熱は、ネッタイシマカ・ヒトスジシマカの吸血によって媒介する感染症です。デング熱は媒介者であるネッタイシマカの生息域と感染地域がほぼ一致しており、全世界で年間約1億人がデング熱を発症し、約25万人がデング出血熱を発症していると考えられています。アジアを中心に熱帯・亜熱帯地域が主な感染地域ですが、昨今の地球温暖化により媒介蚊の生息範囲の拡大が懸念されています。デング熱は軽症ですみますが、デング出血熱という重篤な病型をとると、ショック症状で死亡することもあります。デングウイルスにはI~IV型の4つのタイプがあり、異なるタイプのウイルスに感染する2度目の感染が、デング出血熱を発症するリスクの1つであることが知られています。しかし、2度目の感染でも重症化しない個体がほとんどであり、重症化の原因についてはよく理解されていません。私たちは、タイに居住するデング熱・デング出血熱の患者(15歳以下の子供)を対象として、感染したウイルスのタイプや年齢を調整した上で、デング出血熱に対する感受性または抵抗性を示すヒト遺伝子多型の検出とその進化学的解析を行っています。
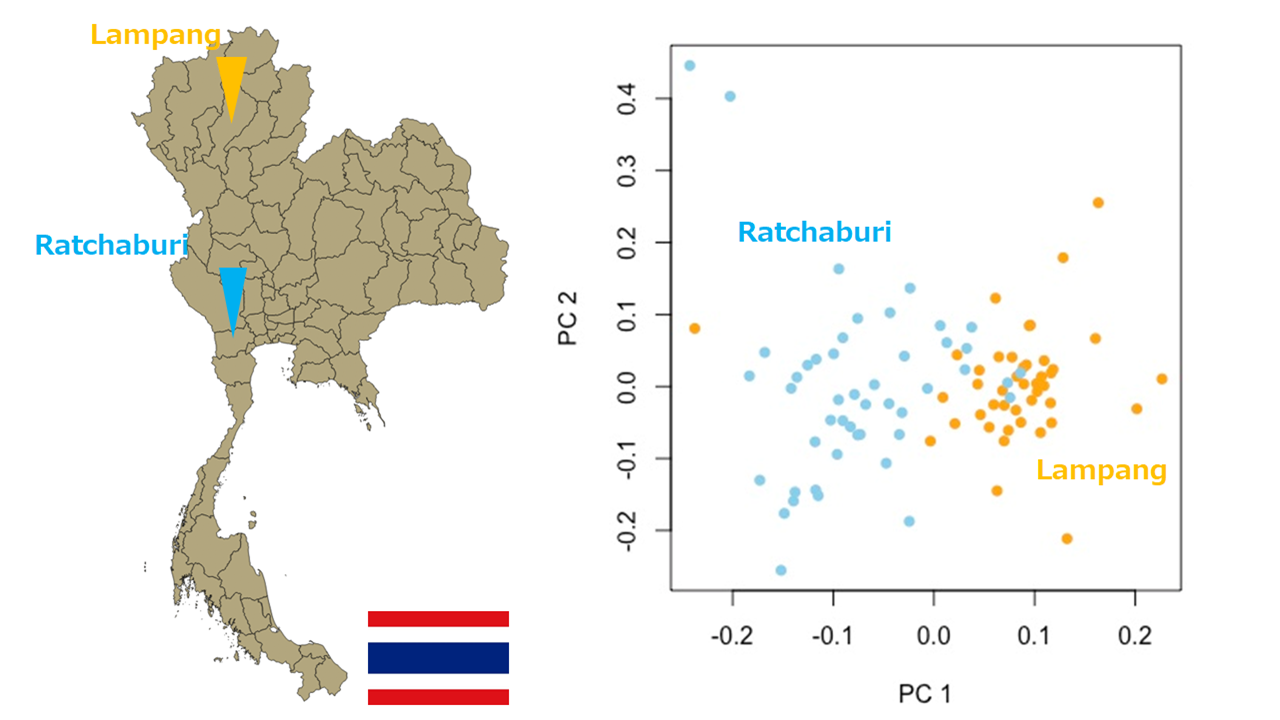
対象はタイのRatchaburi県とLampang県のデング熱感染患者(左図)。集団の階層化が観察されるため(右図)、県を区別して関連解析を行っている。
オセアニア地域集団における体型・脂質代謝関連多型
オーストロネシア語を話すポリネシア人の祖先は、東南アジアを出発し、ニューギニア島を経由してポリネシア地域に拡散したと考えられています。少ない食料からエネルギーをたくさんため込むことができる倹約遺伝子型の形質は、大海原の航海(生存)にとても有利でしたが、現代においては過剰にエネルギーを蓄積し、肥満や生活習慣病などをもたらす要因となったと考えられています。現在のオセアニア地域では、近代化に伴う生活(主に食生活)の変化により、生活習慣病の罹患率が増加しています。私たちは、オセアニア地域の人々を対象に、体型(身長、体重、BMIなど)、脂質(血液中の中性脂肪、コレステロールなど)代謝と関連する多型を、候補遺伝子アプローチによって探索しています。また、データベースを利用して、着目する遺伝子の発現量と関連するSNPも調べています。
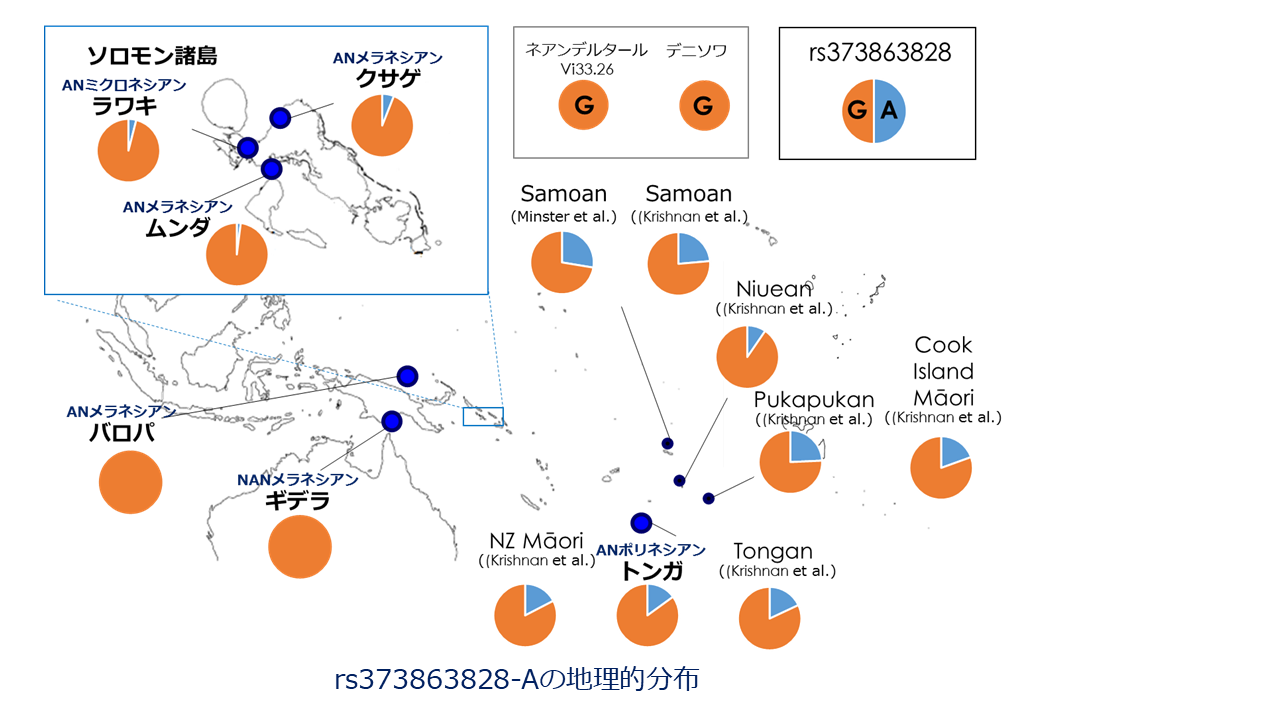
ポリネシアのサモア人やトンガ人でBMIと強く関連するCREBRF遺伝子変異(rs373863828-A)の地理的分布。rs373863828-Aはオセアニア地域特異的に観察され、特に肥満者の割合が高いポリネシア人での頻度が高い。rs373863828-Aに正の自然選択が作用した痕跡がみられることから、ポリネシア人の倹約変異と考えられている。
ゲノムインフォマティクス
様々な生物種のゲノムデータベースが公開されていますが、多様性情報まで含めて考えると最も充実しているのはヒトのデータベースです。ヒトゲノム研究の分野では、データ駆動型の研究が盛んに行われています。私たちは、データベースから取得した情報のみを利用して、旧人(ネアンデルタール人やデニソワ人のゲノム配列が公開されています)との混血によって現代人の祖先に移入し、現代まで維持されてきたヒトゲノム領域を同定し、そのような領域の個体間・集団間の違い、移入変異によってもたらされる機能変化、各領域に作用した正と負の自然選択について調べています。また、全ゲノム関連解析データ、遺伝子発現データ、メチル化領域データなどを統合し、遺伝子発現・メチル化に影響を与える多型についても調べています。
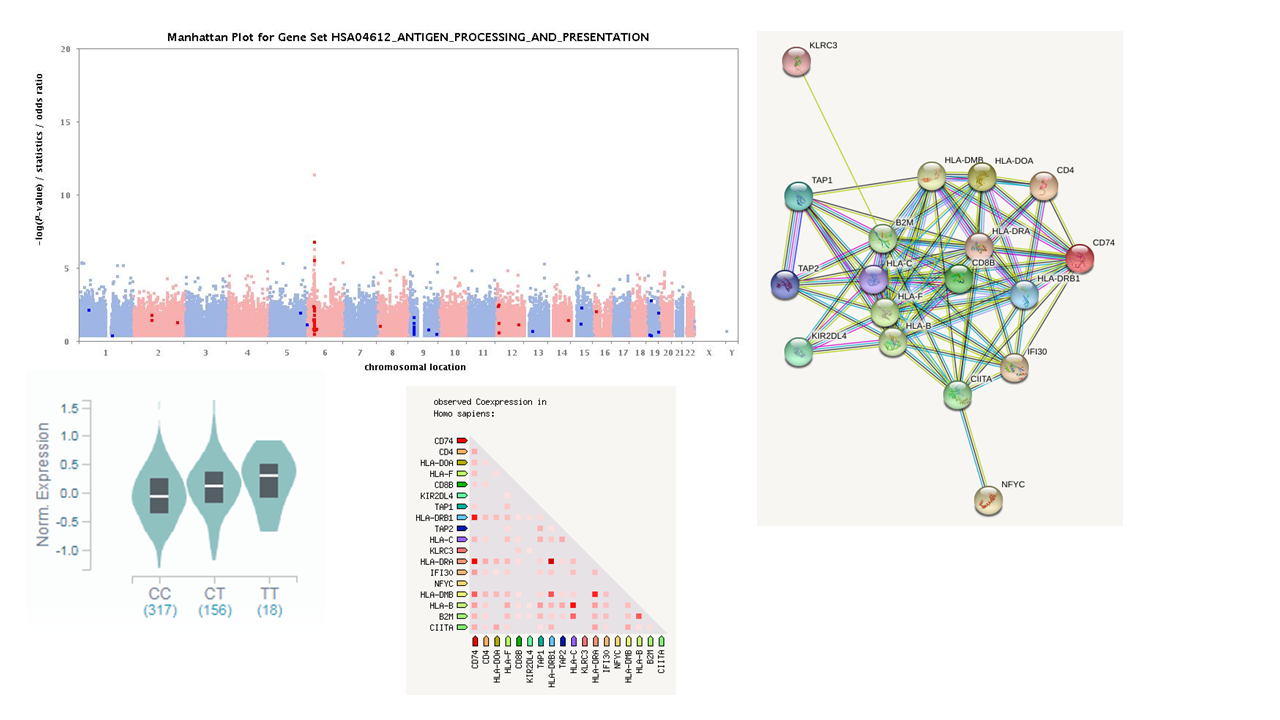
ゲノムワイド関連解析によって有意な関連が観察されたSNPに対して行ったゲノム情報解析結果。データベースのみを利用して、SNPと遺伝子発現量の関連や有意なSNPの近傍に位置する遺伝子群のネットワークを調べることなどができる。
理論集団遺伝学・理論生物学
-集団ゲノム解析-
集団ゲノム学的解析を行うソフトウェアは多数公開されており、データさえあれば何かしらの結果を得ることができます。重要なことは、結果の信頼度を検証することです。私たちは、合祖シミュレーションによって疑似データ(正解データ)を生成して同様の解析を行い、ソフトウェアが正しくモデルを再現できているかを確かめながら研究を進めています。また、実データをもっともうまく説明できる各種パラメタを推定したり、実データの解析に有効な指標の開発も行っています。
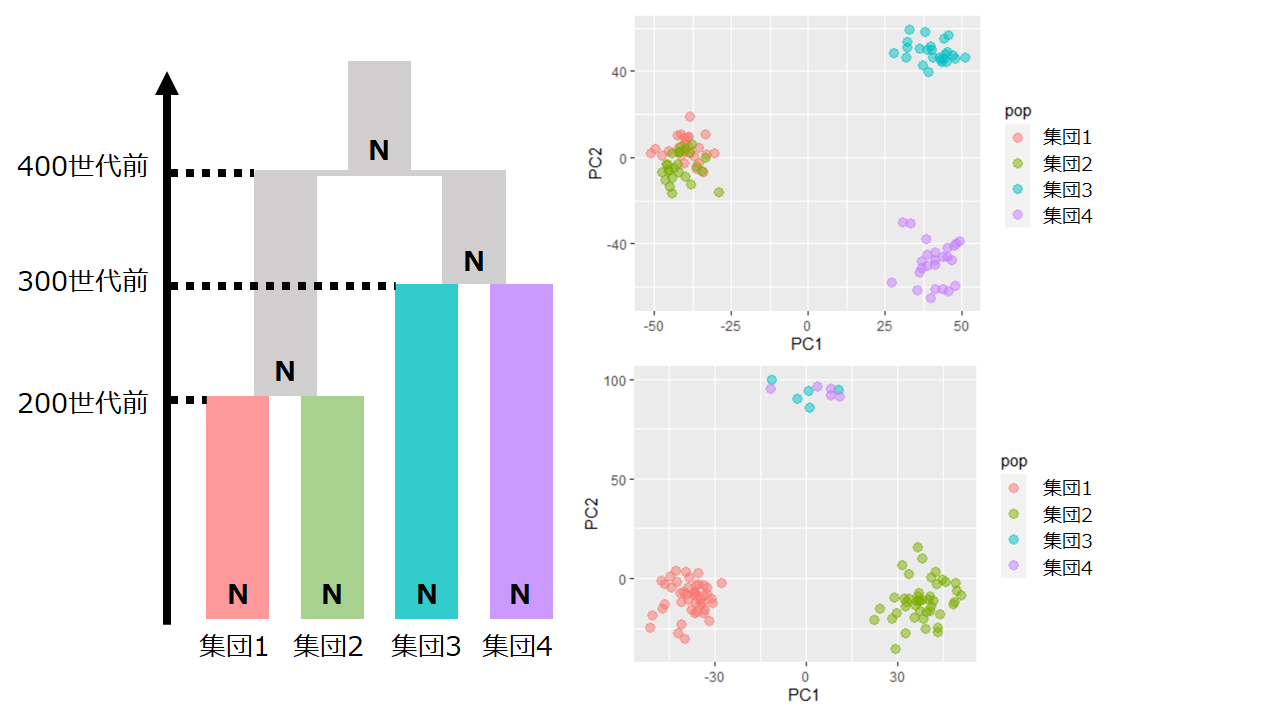
サンプルサイズが主成分分析の結果に与える影響。4集団の進化モデル(左図)を仮定して合祖シミュレーションを行った。右上図は各集団から25人ずつサンプルして主成分分析を行った結果をプロットしたもの。右下図は集団1と集団2からは45人ずつ、集団3と集団4から5人ずつサンプルして主成分分析を行った結果をプロットしたもの。集団1と集団2は遺伝的に近縁となるはずだが、右下の図からは集団1と集団2は遺伝的に遠いと解釈してしまう。サンプルサイズは結果に大きな影響を及ぼすことが理解できる。
-感染症数理モデル-
数理モデルを用いて、(新型コロナウイルス感染症のように)すぐには根絶できないような感染症をコントロールするための有効な対策について考えています。
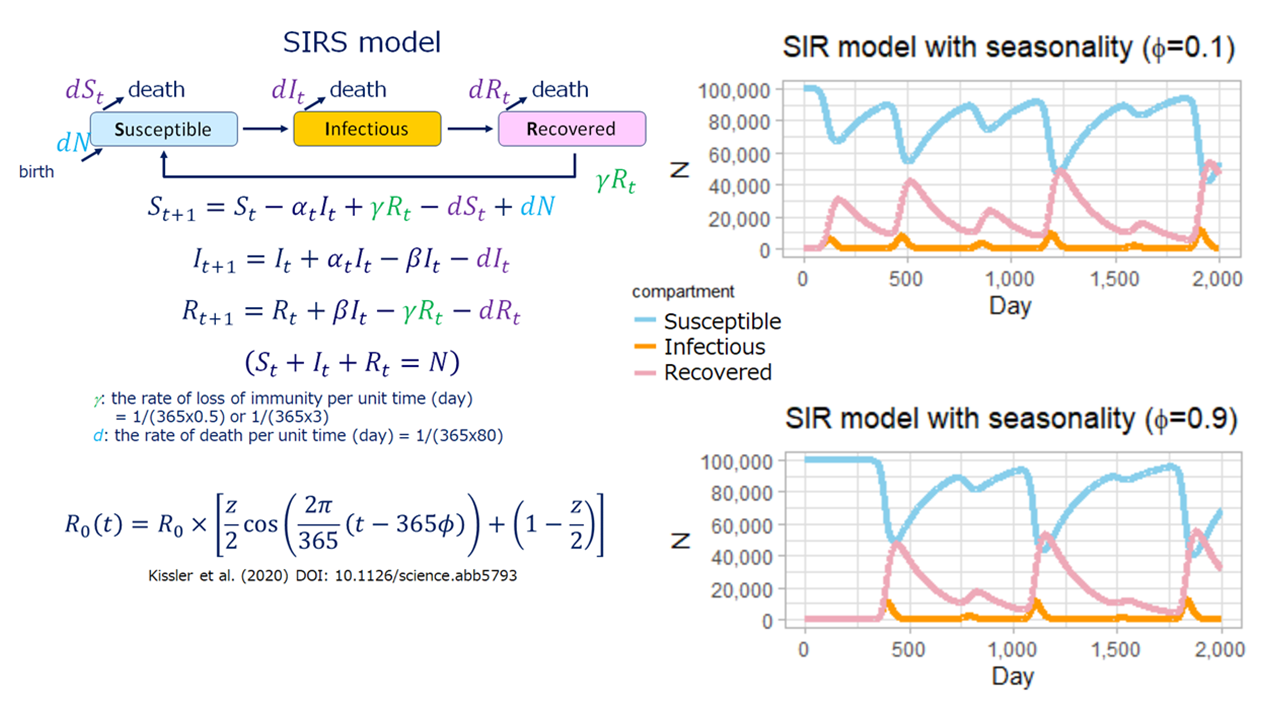
感染症の数理モデルを用いて計算した新型コロナウイルス感染者数の推移。修正免疫を獲得できず(免疫が時間経過に伴い消失する)、1年周期の季節性がある(基本再生産数(R0)が変化する)と仮定したSIRSモデル(左図)。季節性の強さに応じて数年間隔(不定期)で流行が繰り返される(右図上は弱い季節性、右図下は強い季節性を仮定)。